Answer of December 2018
For completion of the online quiz, please visit the HKAM iCMECPD website: http://www.icmecpd.hk/
Clinical History:
This boy has been managed for known short stature since presentation at 6 years of age. He has reached 12 years of age at the time of obtaining these radiographs.
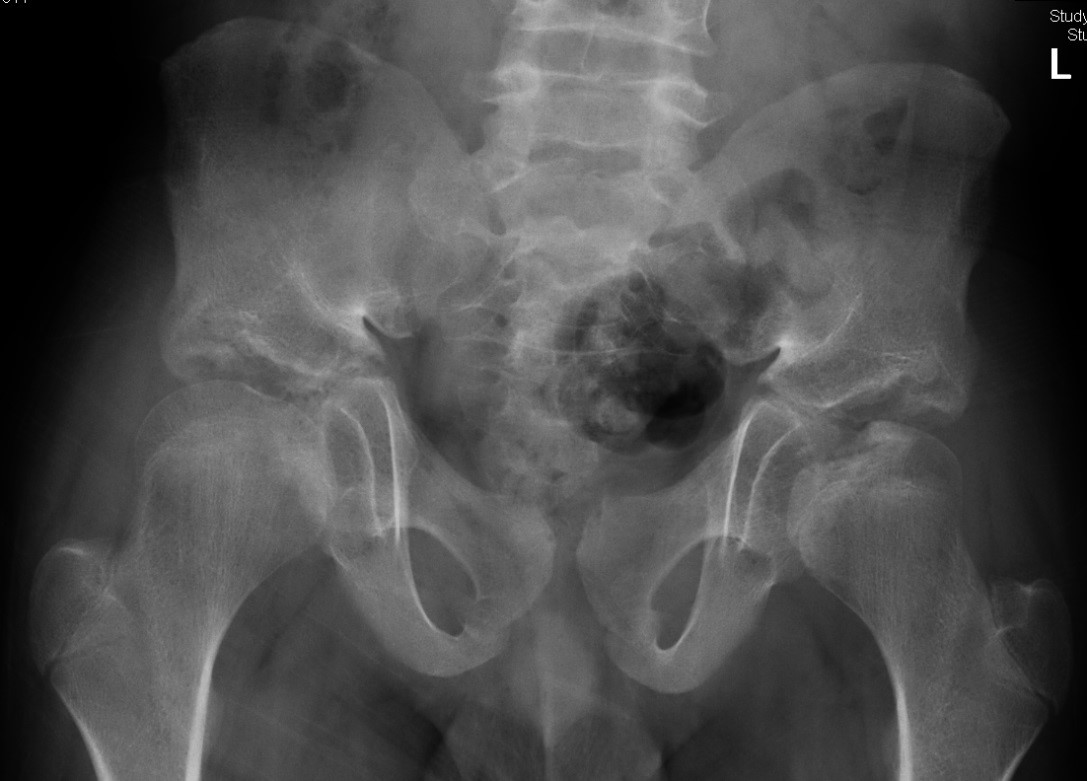
Image 1 of 2
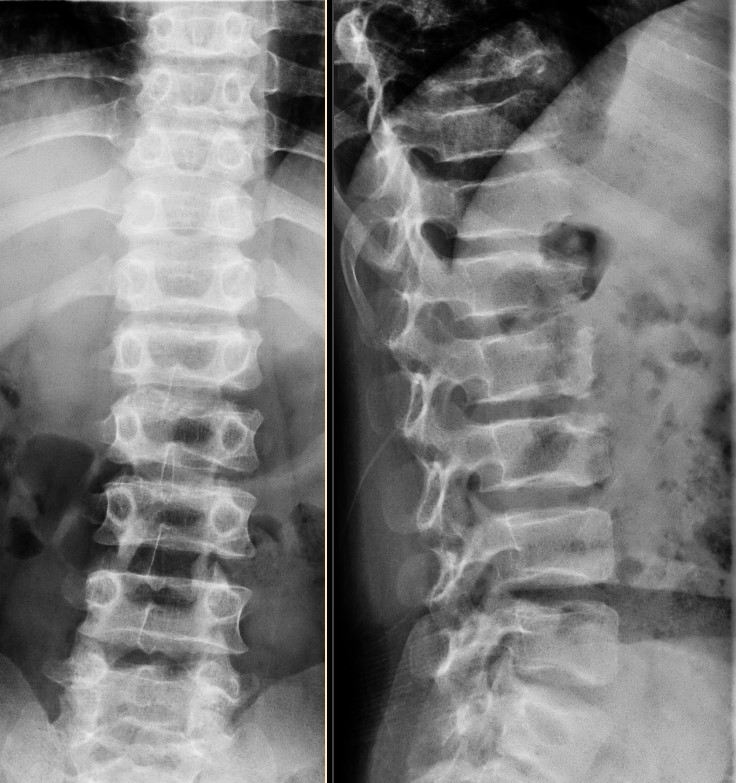
Image 2 of 2
Imaging Findings:
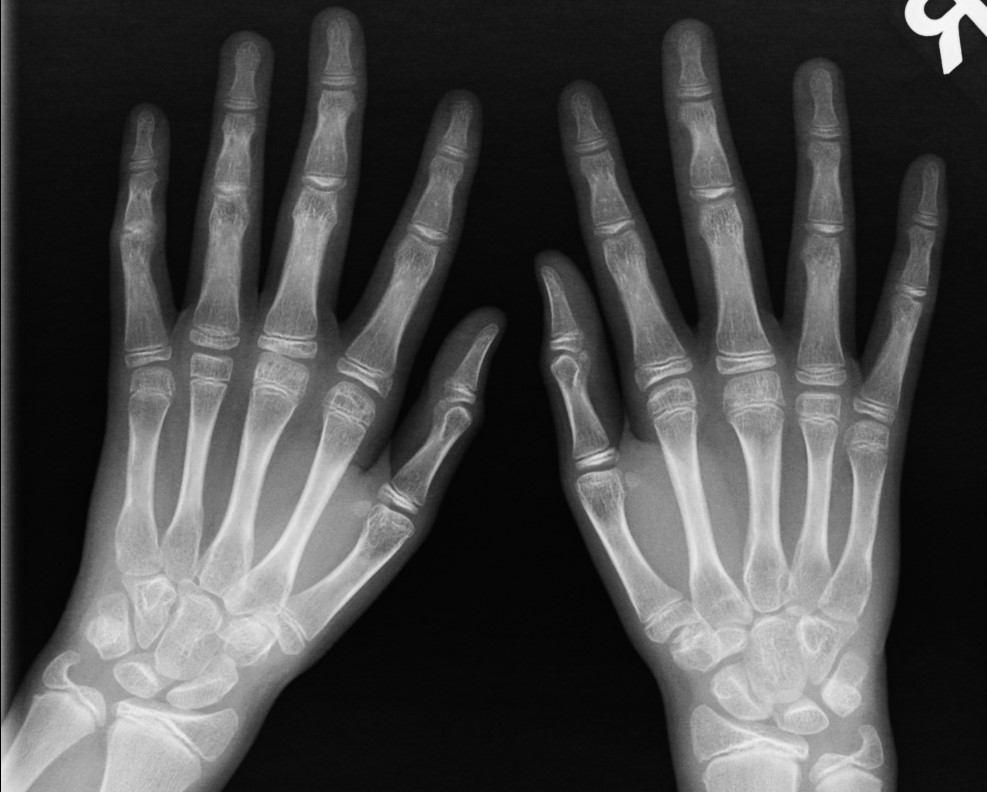
- Multiple irregular and hypoplastic epiphyses involving the femoral heads and metatarsals.
- Small pelvis with underdeveloped iliac ala, clefting of the weightbearing portion of the acetabula.
- Diffuse platyspondyly.
Diagnosis:
Spondyloepiphyseal dysplasia tarda (SEDT)
Discussion:
Spondyloepiphyseal
dysplasia tarda (SEDT) is a skeletal chondrodysplasia characterized by abnormal
development of long bone epiphyses and the spine.
The classical type is X-linked recessive, and exclusive to males. However, more recently autosomal dominant and autosomal recessive modes of inheritance have also been identified. The TRAPPC2 gene is usually involved. It is a gene that encodes a protein known as sedlin, which is responsible for intracellular signaling.
Patients mostly present at the age of 5 to 10 years with short stature or joint pain. (The term ‘tarda’ means presenting late, as compared to the congenita form which presents at birth.) Other physical findings include barrel chest, disproportionately short neck and trunk, disproportionately long arms.
Key radiologic findings include dysplasia of multiple epiphyses, a small pelvis, and underdeveloped acetabula with clefting.
Diffuse platyspondyly is present in virtually all cases, often with kyphosis or scoliosis. Hyperostosis at the posterior two-thirds of the vertebral body giving a hump-shaped appearance is patholgonomonic although not universally present (it is absent in the presented case). Progressive narrowing of the interpedicular distance from superior to inferior is also noted in the autosomal recessive form.
The abnormalities in the hands and feet are usually clinically silent, but subtle underdevelopment of the metacarpal and metatarsal epiphyses can still be detected radiographically (as in the presented case).
The skull is typically spared.
The patients may develop premature osteoarthritis due to underdevelopment and irregularity of the epiphyses, predisposing to incongruity of joint articulations.
The diagnosis is usually made on clinical and radiological grounds. The patients generally do not have biochemical abnormalities on serum or urine analysis. There is no specific medical treatment for SEDT. Orthopaedic intervention may be required for correcting scoliosis or kyphosis and treating complications such as premature osteoarthritis which is most common in the hips.
The peripheral skeletal abnormalities in SEDT are indistinguishable from multiple epiphyseal dysplasia (MED). The distinction between MED and SEDT relies on demonstration of a radiographically normal spine.
The congenital form (spondyloepiphyseal dysplasia congenita, SEDC), which involves a mutation in the COL2A1 encoding for type II collagen, has similar but more exaggerated clinical and radiological features compared with SEDT, and typically presents at birth as a short-trunk and short-limb rhizomelic dwarfism. Epiphyseal ossification is markedly delayed in SEDC.
Morquio Syndrome or type IV mucopolysaccharidosis (MPS) may also present with diffuse platyspondyly and delayed ossification of the epiphyses. However, the hands typically show proximal pointing or rounded tapering of the metacarpals, and underdevelopment of carpal bones are often present. Certain features of dysostosis multiplex found in Hurler’s and other types of MPS including oar-shaped ribs and ping-pong paddle ilium are usually absent in Morquio’s syndrome. Atlantoaxial subluxation is a potentially life-threatening complication in Morquio’s syndrome.